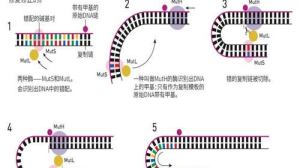
结直肠癌通常为散发病例,但家族聚集现象同样较常见。在所有肠癌患者中,约25%的患者有相应家族史,约10%的患者 […]
部分病友确诊恶性肿瘤后,会非常紧张地问医生两个问题:这个病会传染么?这个病会遗传么? 一般情况下,为了消除病友 […]


 top1
top1 top2
top2 top3
top3 top4
top4 top5
top5





 X
X