
“一个人活着的意义,不能以生命长短作为标准,而应该以生命的质量和厚度来衡量。”得了这个病,活着对我是一种折磨和痛苦。我要有尊严地离开爸爸妈妈,你们要坚强地、微笑着生活,不要为我难过。我走之后,头部可留给医学做研究。希望医学能早日攻克这个难题,让那些因为‘渐冻症’而饱受折磨的人,早日摆脱痛苦……
最近,这份遗嘱,让无数网友泪目。
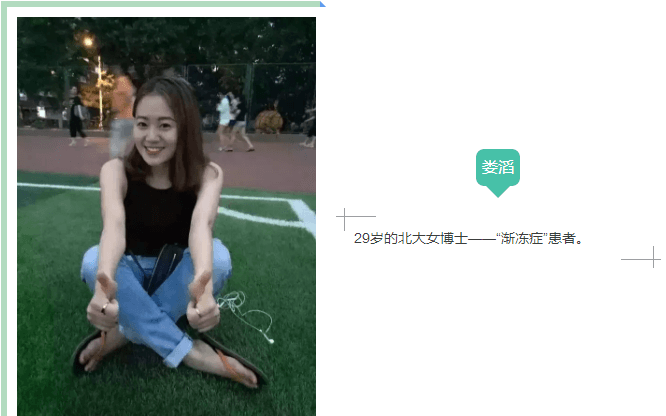
这段感人的文字,是29岁的北大女博士娄滔患上“渐冻症”后,清醒时留下的“遗嘱”。
10月9日清晨7时,娄滔被接到武汉汉阳医院,家属代替她在人体器官捐赠登记表上,签下了名字。
高学历、高颜值,正值大好年华,却患上世上五大绝症之一的渐冻症,娄滔的不幸经历让人扼腕痛惜。
值得献上敬意的是,娄滔有一颗善良的心,她的选择也弥足珍贵。娄滔捐献器官,不仅可带动全社会关注器官捐献事业,也带动更多人关注渐冻症。

对多数人来说,渐冻症是一个陌生的疾病,但它是一个极端折磨人的疾病,比癌症还要残忍的绝症。据统计,从出现症状起,其5年死亡率高达90%。渐冻症患者的渗透就像被冰雪冻住,今天是腿、明天是手,最后连控制眼球转动的肌肉都不例外。
渐冻症的来龙去脉
肌萎缩侧索硬化(ALS)也叫运动神经元病(MND),后一名称英国常用,法国又叫夏科(Charcot)病,而美国也称卢伽雷氏(Lou Gehrig)病。我国通常将肌萎缩侧索硬化和运动神经元病混用,通俗的叫法是“渐冻症”。
它是上运动神经元和下运动神经元损伤之后,导致包括球部(延髓支配的这部分肌肉)、四肢、躯干、胸部腹部的肌肉逐渐无力和萎缩。肌萎缩侧索硬化的病因至今不明。20%的病例可能与遗传及基因缺陷有关,另外有部分环境因素,如遗传、重金属中毒等,都可能造成运动神经元损害。
ALS的出现往往悄无声息。患病初期虽无疼痛,但患者神经细胞支配骨骼肌的能力受到影响。他们会出现一些细微的症状,如走路磕绊、行动笨拙和口齿不清。而这些症状常常会被忽视。
法国神经病学先驱让-马丁·沙尔科(Jean-Martin Charcot)在1869年首次提出并命名该病,所以此病在法国又称为夏科病,通过“肌萎缩侧索硬化”这个名称概括了此病的特点:“肌萎缩”指肌肉缺乏营养;“侧索”是脊髓的一个区域,是死亡的运动神经元所在的位置;“硬化”则是指神经退化过程中的组织硬化或瘢痕化。
因为著名棒球运动员卢·伽雷(Lou Gehrig)的无故掉球和跌倒,这种疾病才走进了公众视野。
卢·伽雷曾被誉为铁马,连续14年征战超过2130场比赛。他于1939年6月被确诊为ALS,一个月后就在扬基体育场悲壮退役。卢·伽雷的病情发展迅速,当年12月就虚弱到无法参加自己入选名人堂的典礼了。随着病势加重,他最终卧床不起,并于1941年结束了年仅37岁的生命。
这也是此病在美国称为卢伽雷氏病的原因。
尽管这是一种致命的疾病,但不知为何,也有约10%的患者生存期达到10年以上。这部分患者中就包括著名物理学家史蒂芬·霍金,他已在ALS的折磨下生活了50余年。霍金先生让更多人了解了渐冻症。

近来的研究显示,环境因素对诱发ALS仅起到很小的作用,可能只是增加了携带易感基因人群的患病风险。
最让人感到困惑的是疾病发生的随机性,只有不足10%的病例是由父母遗传给孩子,其余的病例都是非遗传性或者说散发性的。
2014年,“ALS冰桶挑战”掀起互联网风暴。年轻的波士顿学院棒球队前队长皮特·弗拉泰斯被诊断为ALS,之后他在Facebook上发布视频向他的朋友们发出挑战——把冰水浇在头上,为ALS协会筹集资金。
此举引发了巨大的轰动并得到大量名人的响应,包括马克·扎克伯格、比尔·盖茨、奥普拉·温弗莉、莱昂纳多·迪卡普里奥和勒布朗·詹姆斯在内的众多知名人士都参加了挑战。
短短8周时间,Facebook用户就发布了超过1700万条挑战视频。最终支持者共捐出超过1.15亿美元,其中67%用于科研,20%用于患者和社会服务,9%用于公众及专业教育。
每年,美国有超过6000人确诊为ALS。患病年龄多为50~60岁,但也有患者是在很年轻或者80岁的时候发病的。
发病时,脑部和脊髓的运动神经元会开始死亡,而这些细胞的功能是从大脑经脊髓向肌肉发出信号。因此这将导致患者的活动能力、敏捷程度、语言能力甚至吞咽功能下降。
大多数患者脑部高级认知功能并未受损,只能眼睁睁地看着自己的身体一点点地被疾病吞噬。他们很快就被困在轮椅上,最终卧床不起,然后逐渐丧失交流、进食或呼吸的功能,大多数患者会因呼吸衰竭在3~5年内去世。
FDA唯一批准用于治疗ALS的药物是谷氨酸阻滞剂利鲁唑,平均也只能延长患者3个月的生命,但尚无方法将其治愈。
在过去10年里,精准的测序技术使我们在生物学层面对疾病有了更深的了解。越来越多的研究显示,许多不同的基因都跟这种疾病有关,它们单独表达或共同作用,增加了个体的患病几率。一些基因突变甚至与70%以上的家族性ALS和约10%的散发性ALS相关。
反过来,这些新的基因研究也为更好的治疗指出了方向。
基因沉默技术是某些ALS潜在的治疗手段,作用于特定目标基因的两种药物有望在今年进行临床试验。与此同时,研究人员也在寻找特异性的生物标记物,包括体液或脑电活动中的可测量成分,这将有助于临床医生早期诊断和定量评估疾病进展。这些生物标记物对其他药物的研发也很有帮助。
早期的遗传发现
大多数家族性ALS患者有50%的几率把疾病传给下一代。尽管在患者中只占很小的比例,但他们极大地加深了人类对ALS遗传机制的认知。
关于ALS的第一个遗传学发现,是在1993年确认了SOD1基因突变,约20%的家族性ALS患者都携带有这种突变。SOD1基因负责编码的是抗氧化物超氧化物歧化酶,这种酶的作用是将高反应性超氧化物分子转化为无害的形式。突变导致酶的作用超出了正常范畴,细胞获得了一些新的功能,导致了神经元内某些蛋白形态的改变。
通过对ALS患者进行尸检,研究人员发现绝大多数患者都有一个典型的脑病理特征,那就是运动神经元内的蛋白聚集。
在ALS患者的细胞中,让神经元正常工作的循环体系彻底崩溃。包括酶在内的所有蛋白质,在细胞内合成时都需要保持精准的三维结构才能发挥正常的功能,基因突变则会导致个别蛋白因错误折叠而聚集。出现这种情况时,细胞会通过泛素这一分子标记物,将结构异常的蛋白标记出来并清除掉。但如果清除体系的负荷过重,垃圾就会逐渐堆积。在某些家族性ALS患者的运动神经元中,就能够发现大量泛素标记的SOD1蛋白聚集。
ALS研究领域最重要的里程碑出现在2006年,科学家将目光转移到不携带SOD1突变的ALS患者上,他们发现了另一种被称为TDP-43的蛋白。这种蛋白聚集在几乎每一位患者的运动神经元内。
TDP-43蛋白本来存在于人体的细胞核内,在RNA翻译成蛋白质的过程中发挥重要作用。然而,在ALS患者体内,TDP-43不知为何离开了细胞核,它们聚集在细胞质中,有时还会吸引更多的这类蛋白进入细胞质。
科学家仍在探索,这一过程中的TDP-43究竟是导致了功能缺失(因其离开细胞核)还是毒性功能获得(因其在细胞质中聚集),还是两者兼而有之。
科学家发现大多数患者体内都会出现TDP-43的聚集后,他们便着手研究这种蛋白的基因TARDBP,并在一些遗传家系中发现了罕见突变。这一重要突破从理论上确定了RNA结合蛋白的这一行为可能导致ALS。随后,更多参与RNA调节的蛋白被确定为ALS致病因素,预计还会有更多的发现。
21世纪头十年,ALS的遗传学发现呈现出井喷之势,每年都约有1~2个致病基因被确认,但最振奋人心的发现此时仍未到来。
2011年,两个研究团队分别报道了同一个特殊的突变基因,该基因的名称也同样特殊—C9ORF72。在健康人群中,C9ORF72所包含的一段DNA序列的重复次数为2~23次,但在突变的人群中,这个片段的重复次数可达数百次甚至上千次。随后的研究显示,这些过度重复的序列可以解释40%~50%的家族性ALS病例和5%~10%的散发性病例。
研究人员提出了三种细胞机制来解释这个神秘基因的突变是如何导致ALS的。![]()
通俗的说,DNA重复片段会影响蛋白的合成,导致其功能的降低;重复序列使蛋白无法发挥正常功能,导致了毒性功能获得;当一个人的C9ORF72基因发生突变,扩增的重复序列导致了毒性功能获得。
迄今为止,已有的证据更支持“C9ORF72突变引起ALS是毒性功能获得所致”这样的推断,但尚不清楚的是,RNA聚集和蛋白聚集谁是更关键的致病因素。不过这也不要紧,正在研发的治疗策略将同时阻断突变基因生产RNA和蛋白质的能力。
靶向作用于C9ORF72基因的反义药物有望今年投入人体临床试验。研究人员也为SOD1突变导致的家族性ALS患者设计了反义药物,这项研究的早期临床试验显示鞘注给药是安全的,药物可以通过脑脊液的流动分布在脑周围并进入运动神经元。
寻找突变基因
有明确遗传规律的家族性ALS毕竟只占少数,目前最大的挑战是研究散发性ALS患者,确定是哪些突变,让这些患者的患病风险升高了。目前全世界的科研人员都在积极收集ALS患者的DNA样本,对海量的基因组数据进行分析。
近来多项国际性的研究比对了超过10000例ALS患者和超过20000例正常人,在基因组层面发现了很多差异,这些发现目前都进入了新的研究阶段。新技术也使收集遗传数据变得简单,一天之内完成一个人的全基因组测序已成为现实,而且花费不会超过1000美元。如果只测定所有外显子的序列,会更加便宜快捷。
一旦列出了与ALS易感性相关的基因突变的黑名单,研究人员就可以探索ALS相关遗传突变为什么能够增加患病风险。这些探索包括:
多个基因如何相互作用?
某些形式的ALS是否存在多个突变基因?
环境因素是如何诱发某些患者的疾病的?
生物标记物
越来越多的研究显示,导致ALS的真凶不仅仅是运动神经元的死亡,胶质细胞在其中也起到了重要作用。胶质细胞在脑内和中枢神经系统中数量众多,甚至超过了神经元。它们可以发挥多种功能:有的为神经元提供物理上的支撑,有的可调节脑内环境,特别是神经元和突触周围的液体。
近期关于SOD1突变小鼠的研究又有了惊人的发现:尽管运动神经元中存在毒性SOD1蛋白,但只要阻断胶质细胞中突变基因的合成,就可以延长小鼠的生存期。
运动神经元似乎是疾病的缘起,而前者与胶质细胞之间的信号传导,则促进了疾病的发展。胶质细胞可能也会通过产生毒性因子加剧ALS,但科学家尚不清楚这种因子是什么,也不知道它们是如何发挥作用的。一旦这种(或这些)因子被探明,我们可以阻止其产生,或者阻止它们向运动神经元传递异常信号,这样就能延缓或治愈ALS了。
在破译ALS成因的研究中,科研人员也一直在寻找能够帮助医生评价疾病进展的生物标记物。比如,目前已经有研究试图在易获取的体液(如血液或脑脊液)中检测C9ORF72基因编码的异常重复蛋白,这些检测手段将有助于疾病的早期诊断。其他研究则着眼于开发影像技术,以便在ALS患者脑内的TDP-43蛋白团块攻击运动神经元之前及时检测到它们,所有这些生物标记物都可以作为评价临床试验有效性的标准。
遗传学和基因组学的快速发展以及新的生物标记物的开发,使ALS研究进入了精准医疗的时代。在不远的未来,患者将依据ALS类型分组,接受特定的治疗或预防。
日前有媒体报道,自2017年9月1日起,唯一经严格临床试验并验证可有效延长渐冻症患者生存期的药物—利鲁唑陆续进入全国16个省市的医保乙类药物目录,新政策将极大地降低渐冻症患者就医的负担,患者平均每年治疗费用可节省约41600元。
不让渐冻症患者以一己之力对抗困苦,这是政府部门和全社会的责任。通过医保体制变革,让渐冻症患者感受到制度暖意,他们才更有力量向前。
本文仅供医学药学专业人士阅读













 X
X